使用说明:
1.靶DNA序列的选择和PCR上游引物(即Target-specific DNA oligo)的设计与合成。
a.靶DNA序列的选择。选择的靶DNA序列需要满足如下2个条件。以pUCm-T载体(D2006)作为基因编辑的靶基因为例展示的对于靶DNA序列的选择请参见图4。
(a)选择的靶DNA序列的3'端必须紧邻着PAM序列(NGG)。只有PAM序列上游的20nt DNA序列才能被选择用于CRISPR/Cas9系统的靶序列。
(b)任何紧邻着PAM序列(NGG)的靶序列都可被选用。但为了降低切割时的脱靶概率,整个靶序列(包括PAM序列)与其它任何靶DNA序列之外的基因组DNA序列至少有三个错配碱基。如果错配碱基出现在PAM序列或紧邻着PAM序列,会降低脱靶效应的几率。
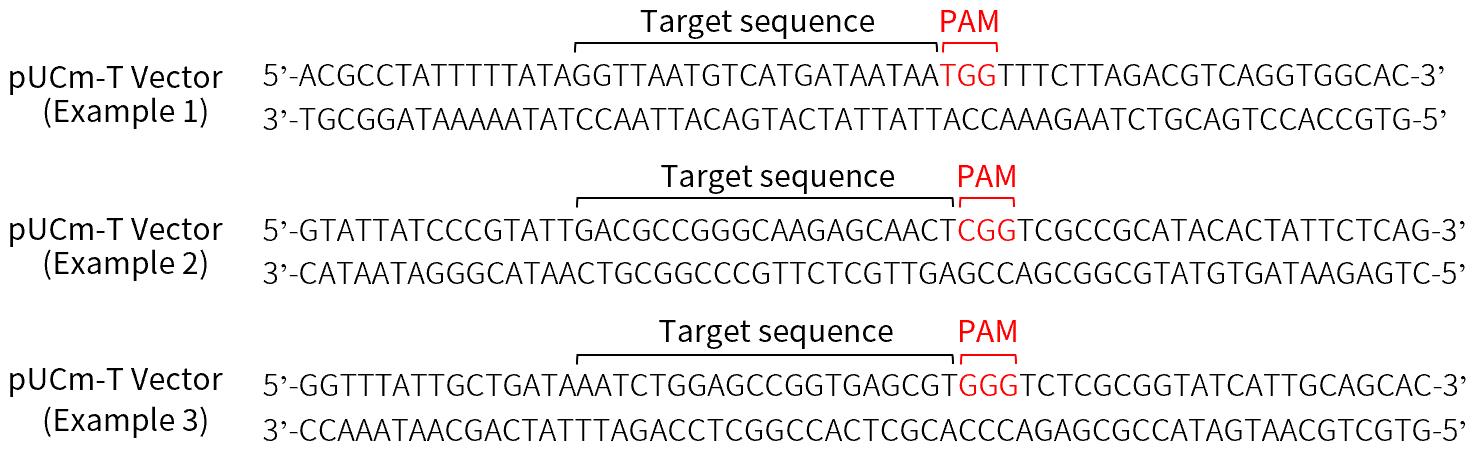 图4. 基于CRISPR/Cas9系统的基因编辑靶DNA序列的选择示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示对于靶DNA序列的选择。
图4. 基于CRISPR/Cas9系统的基因编辑靶DNA序列的选择示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示对于靶DNA序列的选择。
b.PCR上游引物(即Target-specific DNA oligo)的设计与合成。PCR上游引物必须包括如下的4部分序。(a).引物的5'端为T7启动子序列(17nt)加额外的四个保护碱基(4nt)。(b).转录起始位点0~2个G。添加的G的个数依赖于靶序列的5'端。T7启动子至少需要两个G才能有效转录。如果选择的靶序列已经包含2个G,就不需要在转录起始位点添加额外的G。如果包含1个或0个G,就需要相应增加1个或2个额外的G。超过2个G会降低切割效率。(c).20nt长度的sgRNA靶序列。(d).引物3'端14nt长度的Scaffold Template退火序列。以pUCm-T载体(D2006)作为基因编辑的靶基因为例展示的PCR上游引物设计请参见图5。
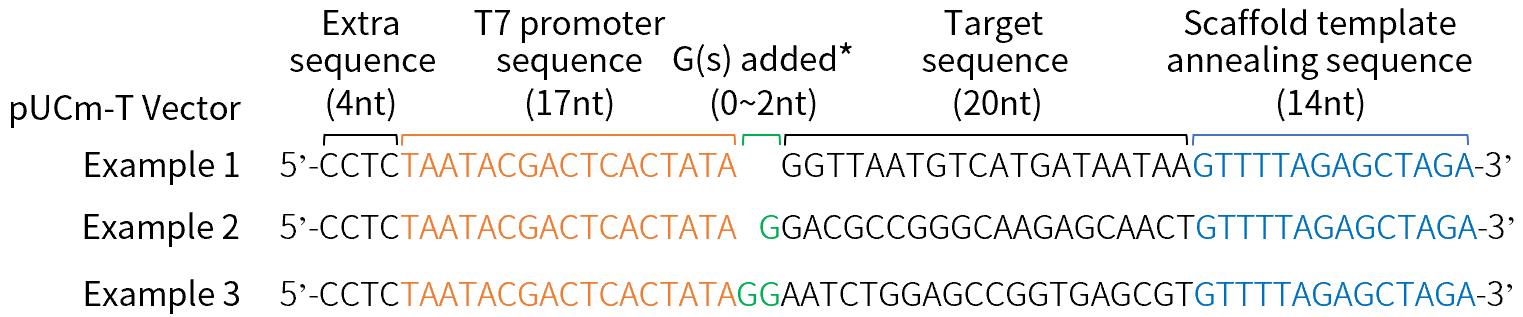 图5. BeyoCRISPR™ Two-Step sgRNA Synthesis Kit (D7083)所需PCR上游引物(即Target-specific DNA oligo)的设计示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示设计的PCR上游引物(即Target-specific DNA oligo)序列。
图5. BeyoCRISPR™ Two-Step sgRNA Synthesis Kit (D7083)所需PCR上游引物(即Target-specific DNA oligo)的设计示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示设计的PCR上游引物(即Target-specific DNA oligo)序列。
2.PCR扩增合成sgRNA转录所需DNA模板。
a.溶解PCR反应所需的各种溶液。
b.参考下表在冰浴上配制PCR反应体系。如果有多个类似的PCR反应,可以先把下表中除了自行设计合成的Target-specific DNA Oligo之外的所有组分提前预混合,然后分装到各PCR反应管内,再在各管内加入Target-specific DNA Oligo。
注:由于此步骤产生的PCR产物后续要作为模板产生sgRNA,所以需要严格按照RNA操作的规范进行,避免RNase污染,相关试剂和耗材需要确保是RNase free的,或者是经过DEPC处理已经去除RNase的。
c.用移液器轻轻吹打混匀或轻微Vortex混匀,室温离心数秒,使液体积聚于管底。
d.如果PCR仪没有热盖,则在PCR管内滴入一滴矿物油(mineral oil, ST275)。
e.将设置好的PCR反应管置于PCR仪上,开始PCR反应。PCR反应参数的设置可以参考如下表格。
f.PCR反应结束后,可以取约3-5µl PCR产物用于2%或3%的DNA琼脂糖凝胶电泳分析。检测PCR扩增产物条带的单一性和大致产量。如果条带比较单一,产量也符合预期(预期20µl PCR产物中约含有8µg模板DNA),就可以进行后续步骤了。如果产量略偏低,后续的sgRNA的产量也会相应下降。
3.体外转录获得sgRNA。
a.溶解并混匀转录反应所需的各种溶液,将T7 RNA polymerase置于冰浴上或冰盒内。
b.参考下表在冰浴上配制如下转录反应体系。如果同时进行多个转录反应,可以把下表中除sgRNA Template之外的所有组分提前预混合,然后分装到各反应管内,再在各管内加入sgRNA Template。
注:由于涉及RNA操作,需要严格按照RNA操作的规范进行,避免RNase污染,相关试剂和耗材需要确保是RNase free的,或者是经过DEPC处理已经去除RNase的。可以适当通过按比例放大反应体系,以获得更多的sgRNA产量。放大反应体系对sgRNA的得率基本没有影响。
c.按照上表配制好反应体系后,轻轻混匀(可以用移液器吹打混匀或用Vortex在最低速度轻轻混匀),瞬时离心。
d.反应条件:37℃孵育0.5~4h。通常反应时间越长,生成的sgRNA越多。
e.终止反应:70℃孵育10min以终止反应。
4.sgRNA的纯化。
a.取20μl转录反应产物,加入2μl DNase I,37℃孵育15min以充分降解DNA模板。
b.后续推荐使用如下的酚氯仿抽和乙醇沉淀方法进行sgRNA的纯化。也可以使用碧云天生产的R0028 RNAeasy™动物小RNA抽提试剂盒(离心柱式)进行sgRNA的纯化,但该试剂盒每个纯化柱的抽提容量相对较小一点,不能满足10-40µg sgRNA的纯化要求,但可以用于少量sgRNA快速纯化和分析鉴定。
c.在上述反应体系中加入160μl Ultrapure Water,再加入20μl 3M NaAc, pH5.2 (约200μl最终体积的1/10体积),混匀。
d.加入200μl水饱和酚/氯仿混合溶液(等体积),充分Vortex混匀以变性蛋白。
e.4℃ 12000-14000g离心10min。收集上层液体至新的1.5ml离心管(Nuclease free),加入2倍体积无水乙醇,混匀,-20℃沉淀3h或-80℃沉淀1h (如果想得到更高产量的sgRNA,可以考虑沉淀过夜)。
f.4℃ 12000-14000g离心10min。去除上清,加入1ml 70%乙醇。
g.4℃ 12000-14000g离心5min。去除上清,尽量洗净残留液体,打开管盖,室温放置2-5min挥发残留乙醇。
h.加入20μl Ultrapure Water溶解沉淀,-80℃冻存。sgRNA的浓度可以通过测定紫外吸收,以及通过电泳染色进行确定。通常一个反应体系可以获得约10-40μg sgRNA。
5.(选做) 所设计的sgRNA的效果鉴定。
a.参考下表在冰浴上配制如下反应体系。D0511 Cas9 Nuclease (SpCas9)可以向碧云天购买。
b.反应条件:37℃孵育15min。反应时间也可以根据具体的实验目的适当延长至例如30-120min。
c.终止反应:加入1µl Proteinase K (Proteinase K, ST533),混匀,室温孵育10min。
d.电泳检测:取出10μl反应液,加入2μl DNA上样缓冲液(6X) (D0071)进行琼脂糖凝胶电泳,电泳结束后使用D0130 NA-Red (EB升级换代产品)室温染色15min,并使用凝胶成像系统观察实验结果。
e.结果分析:对于设计的不同的sgRNA,可以根据电泳结果中对于底物DNA的切割效率判定sgRNA的设计效果。
常见问题:
1.sgRNA获得量低。
a.模板DNA质量问题:DNA中残留的乙醇、酚、氯仿等有机溶剂会影响转录效率。解决方法:重新制备高纯度的DNA模板。
b.RNA酶污染:RNA酶可以由模板DNA或者操作中带入。RNA酶会降解转录产物,从而使获得的sgRNA量减少。
c.模板DNA量不足:每个sgRNA转录体系需要约2μg DNA模板。转录前需要定量DNA模板并且电泳检测是否条带单一、清晰。使用更大量的DNA模板对于提高sgRNA产量有一定帮助。
2.sgRNA电泳条带模糊、两条带、大小不正确。sgRNA为100nt左右的单链RNA。在水溶液中容易形成二级结构。因此电泳时位置会发生变化。采用变性后快速冷却处理或者甲酰胺变性凝胶电泳可以显著改善电泳效果。
3.sgRNA变性琼脂糖凝胶电泳出现显著小于100nt的弥散带。RNA酶污染造成sgRNA降解。解决方法:采用高质量的无核酸酶酶污染的耗材进行实验。实验过程中注意防止核酸酶污染。
参考文献:
1.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Cell. 2014.156(5):935-49.
2.Marangi M, Pistritto G. Front Pharmacol. 2018. 9:396.
相关产品:
1.靶DNA序列的选择和PCR上游引物(即Target-specific DNA oligo)的设计与合成。
a.靶DNA序列的选择。选择的靶DNA序列需要满足如下2个条件。以pUCm-T载体(D2006)作为基因编辑的靶基因为例展示的对于靶DNA序列的选择请参见图4。
(a)选择的靶DNA序列的3'端必须紧邻着PAM序列(NGG)。只有PAM序列上游的20nt DNA序列才能被选择用于CRISPR/Cas9系统的靶序列。
(b)任何紧邻着PAM序列(NGG)的靶序列都可被选用。但为了降低切割时的脱靶概率,整个靶序列(包括PAM序列)与其它任何靶DNA序列之外的基因组DNA序列至少有三个错配碱基。如果错配碱基出现在PAM序列或紧邻着PAM序列,会降低脱靶效应的几率。
图4. 基于CRISPR/Cas9系统的基因编辑靶DNA序列的选择示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示对于靶DNA序列的选择。b.PCR上游引物(即Target-specific DNA oligo)的设计与合成。PCR上游引物必须包括如下的4部分序。(a).引物的5'端为T7启动子序列(17nt)加额外的四个保护碱基(4nt)。(b).转录起始位点0~2个G。添加的G的个数依赖于靶序列的5'端。T7启动子至少需要两个G才能有效转录。如果选择的靶序列已经包含2个G,就不需要在转录起始位点添加额外的G。如果包含1个或0个G,就需要相应增加1个或2个额外的G。超过2个G会降低切割效率。(c).20nt长度的sgRNA靶序列。(d).引物3'端14nt长度的Scaffold Template退火序列。以pUCm-T载体(D2006)作为基因编辑的靶基因为例展示的PCR上游引物设计请参见图5。
图5. BeyoCRISPR™ Two-Step sgRNA Synthesis Kit (D7083)所需PCR上游引物(即Target-specific DNA oligo)的设计示例。以碧云天的pUCm-T载体(D2006)作为基因编辑的靶基因为例展示设计的PCR上游引物(即Target-specific DNA oligo)序列。2.PCR扩增合成sgRNA转录所需DNA模板。
a.溶解PCR反应所需的各种溶液。
b.参考下表在冰浴上配制PCR反应体系。如果有多个类似的PCR反应,可以先把下表中除了自行设计合成的Target-specific DNA Oligo之外的所有组分提前预混合,然后分装到各PCR反应管内,再在各管内加入Target-specific DNA Oligo。
| Reagent | Volume |
| Template Mix (2X) | 10µl |
| Target-specific DNA Oligo (10µM) | 1µl |
| Ultrapure Water | 8.5µl |
| BeyoFusion™ DNA Polymerase | 0.5µl |
| Total Volume | 20µl |
c.用移液器轻轻吹打混匀或轻微Vortex混匀,室温离心数秒,使液体积聚于管底。
d.如果PCR仪没有热盖,则在PCR管内滴入一滴矿物油(mineral oil, ST275)。
e.将设置好的PCR反应管置于PCR仪上,开始PCR反应。PCR反应参数的设置可以参考如下表格。
| PCR Step | Temperature | Time | Cycles |
| STEP1 (Initial Denaturation) | 92℃ | 3min | 1 |
| STEP2 (Denaturation) | 92℃ | 30s | 30 |
| STEP3 (Annealing) | 55℃ | 30s | |
| STEP4 (Extension) | 68℃ | 30s | |
| STEP5 (Final Extension) | 68℃ | 10min | 1 |
| STEP6 (Hold) | 4℃ | forever | - |
3.体外转录获得sgRNA。
a.溶解并混匀转录反应所需的各种溶液,将T7 RNA polymerase置于冰浴上或冰盒内。
b.参考下表在冰浴上配制如下转录反应体系。如果同时进行多个转录反应,可以把下表中除sgRNA Template之外的所有组分提前预混合,然后分装到各反应管内,再在各管内加入sgRNA Template。
| Reagent | Volume |
| sgRNA Synthesis Buffer (2X) | 10µl |
| sgRNA Template (PCR Product) | 5µl |
| RNase Inhibitor (40U/µl) | 1µl |
| T7 RNA Polymerase | 1µl |
| Ultrapure Water | 3µl |
| Total Volume | 20µl |
c.按照上表配制好反应体系后,轻轻混匀(可以用移液器吹打混匀或用Vortex在最低速度轻轻混匀),瞬时离心。
d.反应条件:37℃孵育0.5~4h。通常反应时间越长,生成的sgRNA越多。
e.终止反应:70℃孵育10min以终止反应。
4.sgRNA的纯化。
a.取20μl转录反应产物,加入2μl DNase I,37℃孵育15min以充分降解DNA模板。
b.后续推荐使用如下的酚氯仿抽和乙醇沉淀方法进行sgRNA的纯化。也可以使用碧云天生产的R0028 RNAeasy™动物小RNA抽提试剂盒(离心柱式)进行sgRNA的纯化,但该试剂盒每个纯化柱的抽提容量相对较小一点,不能满足10-40µg sgRNA的纯化要求,但可以用于少量sgRNA快速纯化和分析鉴定。
c.在上述反应体系中加入160μl Ultrapure Water,再加入20μl 3M NaAc, pH5.2 (约200μl最终体积的1/10体积),混匀。
d.加入200μl水饱和酚/氯仿混合溶液(等体积),充分Vortex混匀以变性蛋白。
e.4℃ 12000-14000g离心10min。收集上层液体至新的1.5ml离心管(Nuclease free),加入2倍体积无水乙醇,混匀,-20℃沉淀3h或-80℃沉淀1h (如果想得到更高产量的sgRNA,可以考虑沉淀过夜)。
f.4℃ 12000-14000g离心10min。去除上清,加入1ml 70%乙醇。
g.4℃ 12000-14000g离心5min。去除上清,尽量洗净残留液体,打开管盖,室温放置2-5min挥发残留乙醇。
h.加入20μl Ultrapure Water溶解沉淀,-80℃冻存。sgRNA的浓度可以通过测定紫外吸收,以及通过电泳染色进行确定。通常一个反应体系可以获得约10-40μg sgRNA。
5.(选做) 所设计的sgRNA的效果鉴定。
a.参考下表在冰浴上配制如下反应体系。D0511 Cas9 Nuclease (SpCas9)可以向碧云天购买。
| Reagent | Volume |
| Ultrapure water | 20µl |
| Cas9 Reaction Buffer (10X) | 3µl |
| 1µM Cas9 Nuclease (SpCas9) (D0511) | 1µl |
| 300nM sgRNA | 3µl (30nM final) |
| Volume | 27µl |
| Pre-incubate for 10min at 25℃ | |
| 30nM substrate DNA | 3µl (3nM final) |
| Total volume | 30µl |
c.终止反应:加入1µl Proteinase K (Proteinase K, ST533),混匀,室温孵育10min。
d.电泳检测:取出10μl反应液,加入2μl DNA上样缓冲液(6X) (D0071)进行琼脂糖凝胶电泳,电泳结束后使用D0130 NA-Red (EB升级换代产品)室温染色15min,并使用凝胶成像系统观察实验结果。
e.结果分析:对于设计的不同的sgRNA,可以根据电泳结果中对于底物DNA的切割效率判定sgRNA的设计效果。
常见问题:
1.sgRNA获得量低。
a.模板DNA质量问题:DNA中残留的乙醇、酚、氯仿等有机溶剂会影响转录效率。解决方法:重新制备高纯度的DNA模板。
b.RNA酶污染:RNA酶可以由模板DNA或者操作中带入。RNA酶会降解转录产物,从而使获得的sgRNA量减少。
c.模板DNA量不足:每个sgRNA转录体系需要约2μg DNA模板。转录前需要定量DNA模板并且电泳检测是否条带单一、清晰。使用更大量的DNA模板对于提高sgRNA产量有一定帮助。
2.sgRNA电泳条带模糊、两条带、大小不正确。sgRNA为100nt左右的单链RNA。在水溶液中容易形成二级结构。因此电泳时位置会发生变化。采用变性后快速冷却处理或者甲酰胺变性凝胶电泳可以显著改善电泳效果。
3.sgRNA变性琼脂糖凝胶电泳出现显著小于100nt的弥散带。RNA酶污染造成sgRNA降解。解决方法:采用高质量的无核酸酶酶污染的耗材进行实验。实验过程中注意防止核酸酶污染。
参考文献:
1.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Cell. 2014.156(5):935-49.
2.Marangi M, Pistritto G. Front Pharmacol. 2018. 9:396.
相关产品:
| 产品编号 | 产品名称 | 包装 |
| D7081S | BeyoCRISPR™ One-Step sgRNA Synthesis Kit | 20次 |
| D7081M | BeyoCRISPR™ One-Step sgRNA Synthesis Kit | 100次 |
| D7083S | BeyoCRISPR™ Two-Step sgRNA Synthesis Kit | 20次 |
| D7083M | BeyoCRISPR™ Two-Step sgRNA Synthesis Kit | 100次 |
| D0511S | Cas9 Nuclease (SpCas9) | 50pmol |
| D0511M | Cas9 Nuclease (SpCas9) | 250pmol |
| D0511L | Cas9 Nuclease (SpCas9) | 1000pmol |
| D0508S | 基因组编辑突变检测试剂盒 | 25次 |
| D0508M | 基因组编辑突变检测试剂盒 | 100次 |
| D7080S | T7 Endonuclease I (CRISPR等基因突变鉴定用) | 250U |
| D7080M | T7 Endonuclease I (CRISPR等基因突变鉴定用) | 1250U |
| D7080L | T7 Endonuclease I (CRISPR等基因突变鉴定用) | 5000U |
| D7062 | SP6 RNA Polymerase | 500U |
| D7066 | T3 RNA Polymerase | 500U |
| D7069 | T7 RNA Polymerase | 1000U |
| R0102-2kU | RNase Inhibitor | 2000U |
| R0102-10kU | RNase Inhibitor | 10000U |
| R0102-50kU | RNase Inhibitor | 50000U |
| D7073 | DNase I | 200U |
| D7076 | DNase I | 1000U |
| ST532 | Proteinase K (20mg/ml) | 0.2ml |
| ST533 | Proteinase K (20mg/ml) | 1ml |
| ST876-100ml | BeyoPure™ Ultrapure Water (DNase/RNase-Free, Sterile) | 100ml |
| ST876-500ml | BeyoPure™ Ultrapure Water (DNase/RNase-Free, Sterile) | 500ml |